The team, based at Tohoku University, published their results on June 8 in the International Journal of Molecular Sciences. The treatment candidate has been declared safe by Japan's governing board, and the researchers plan to begin clinical trials in humans in the next year.
"There are currently no disease-modifying therapeutics for neurodegenerative disorders such as Alzheimer's disease, Lewy body dementia, Huntington disease and frontotemporal dementia in the world," said paper author Kohji Fukunaga, professor emeritus in Tohoku University's Graduate School of Pharmaceutical Sciences. "We discovered the novel, disease-modifying therapeutic candidate SAK3, which, in our studies, rescued neurons in most protein-misfolding, neurodegenerative diseases."
In a previous study, the team found that the SAK3 molecule - the base structure of which is found in the enhancement of T-type Ca2+ channel activity - appeared to help improve memory and learning in a mouse model of Alzheimer's disease.
According to previous studies, SAK3 enhances the function of a cell membrane channel thereby promoting neuronal activity in the brain. Typically, SAK3 promotes neurotransmitter releases of acetylcholine and dopamine that are significantly reduced in Alzheimer's disease and Lewy body dementia. The Ca2+ channel enhancement is thought to trigger a change from resting to active in neuronal activity. When the Ca2+ channel is dysregulated in the brain, the acetylcholine and dopamine releases are reduced. The result is a dysregulated system that a person experiences as cognitive confusion and uncoordinated motor function.
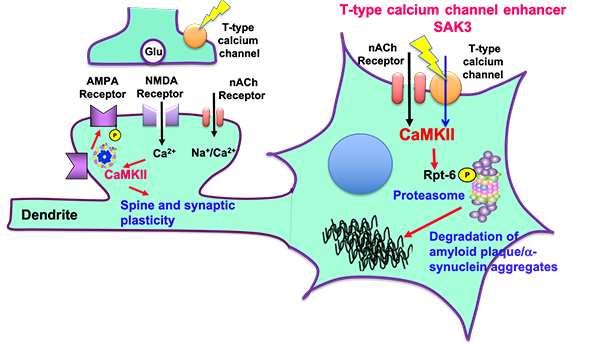
Neurodegenerative diseases such as Alzheimer's disease and dementia with Lewy body (DLB) are caused by the accumulation of aggregated amyloid beta and α-synucelin, respectively. The aggregated proteins impaired the proteasome activity, thereby exacerbating neuronal death. On the other hand, we developed a novel T-type calcium channel enhancer SAK3 in 2017. Since T-type calcium channel is critical for neurotransmitter release, SAK3 enhanced the acetylcholine release in the brain thereby improving learning and memory. We here found that calcium entry through T-type calcium channel activates protein kinase (CaMKII), thereby promoting Rpt-6 phosphorylation. The Rpt-6 phosphorylation promoted the degradation of aggregated amyloid beta and α-synucelin in neurons. This is the first disease modifying therapeutics in most neurodegenerative diseases such as Alzheimer's disease, dementia with Lewy body (DLB), Huntington disease (HD) and frontotemporal dementia (FTD). © Kohji Fukunaga
SAK3 directly binds to the subunit of this channel, resulting in the enhancement of neurotransmission thereby improving cognitive deficits. The researchers found that the same process also appeared to work in a mouse model of Lewy body dementia, which is characterized by a build-up of proteins known as Lewy bodies.
"Even after the onset of cognitive impairment, SAK3 administration significantly prevented the progression of neurodegenerative behaviors in both motor dysfunction and cognition," Fukunaga said.
In comparison, Aduhelm, the Alzheimer's drug recently approved by the U.S. Food and Drug Administration, reduces the number of amyloid plaques in the brain, but it is not yet known if the amyloid reduction actually prevents further cognitive or motor decline in patients. According to Fukunaga, SAK3 helps destroy amyloid plaque - at least in mice.
SAK3 also helps manage the destruction of misfolded alpha-synuclein. Normal alpha-synuclein helps regulate neurotransmitter transmission in the brain. The protein can misfold and aggregate, contributing to what researchers suspect may be an underlying cause of neurodegenerative symptoms. This aggregation can also lead to the loss of dopamine neurons, which help with learning and memory.
"We found that chronic administration of SAK3 significantly inhibited the accumulation of alpha-synuclein in the mice," Fukunaga said, noting that the mice received a daily oral dose of SAK3.
According to Fukunaga, SAK3 enhances the activity of the system that identifies and destroys misfolded proteins. In neurodegenerative diseases, this system is often dysfunctional, leaving misfolded proteins to muck up the cell's machinery.
"SAK3 is the first compound targeting this regulatory activity in neurodegenerative disorders," Fukunaga said. "SAK3 administration promotes the destruction of misfolded proteins, meaning the therapeutic has the potential to solve the problems of diverse protein misfolding diseases such as Parkinson's disease, Lewy body dementia and Huntington disease, in addition to Alzheimer's disease."
Contact:
Kohji Fukunaga


