Повышенное общее укорочение теломер прогнозирует смертность и развитие возрастных болезней в пациентов с наследственным синдромом теломер, а также в обычных когортах людей. Тем не менее генетически обусловленные вариации процесса поддержания длины теломер повышают или снижают риск развития или прогрессии злокачественных опухолей в высокоспецифичной для определенного типа рака манере. Поддержание длины теломер определяется генетическими факторами и обуславливается совместным действием негенетических воздействий на протяжении всей жизни, при этом указанные два основных компонента могут взаимодействовать между собой. Это и другие недавние открытия подчеркивают причинную и усиливающую роли, принадлежащие укорочению теломер в развитии болезней человека.
Теломеры представляют собой локализующиеся на концах хромосом динамичные комплексы с высокой точностью регулирования, представляющие собой участки из тандемно повторяющихся коротких ДНК повторов и ассоциированных с ними белков (рисунок 1) (1).
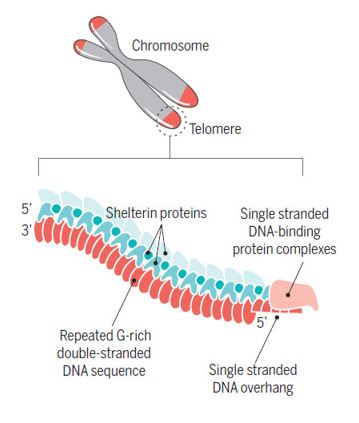
Рисунок 1. Структура теломеры. Теломерный комплекс человека состоит из локализованной на терминальном участке хромосомы последовательности из тандемно повторяющихся последовательностей ДНК, связанных защитными шелтериновыми компонентами белков и дополнительными защитными белками, нависающими над одноцепочечным концевым фрагментом теломерного повтора ДНК. Это упрощенное схематичное изображение не раскрывает детали белковой структуры и архитектуру теломерного комплекса.
Теломеры защищают геномную ДНК с помощью различных механизмов. Одна из выполняемых ими функций заключается в предотвращении распознавания концевых фрагментов линейной хромосомной ДНК как поврежденных концов. Это предупреждает запуск процессов, таких как соединение концевых участков ДНК, рекомбинация ДНК или восстановление повреждений ДНК, которые могут вызывать нестабильность хромосом. Основная система репликации ДНК не может полностью копировать последовательность ДНК до самых концов линейных хромосом. В ходе клеточного деления это приводит к укорочению концевых участков хромосом. У эукариот этот дефект может быть устранен клеточным рибонуклеопротеиновым ферментов теломеразой, добавляющей теломерные повторяющиеся последовательности к концам хромосом, таким образом удлиняя их, компенсируя предшествующее укорочение (2).
Другие вызывающие повреждения механизмы также могут способствовать процессам укорочения теломер. К ним относятся активность нуклеазы, химические (такие как окислительные) повреждения, а также стресс репликации ДНК. В качестве компенсации этих различных процессов теломераза или рекомбинация теломерных повторов могут действовать для увеличения длины теломер (3).
Во многих типах клеток человека уровни теломеразы (или ее активности в отношении теломер) ограничены и теломеры человека укорачиваются на протяжении всей жизни. Степень укорочения примерно пропорциональна рискам развития распространенных, часто сочетанных болезней старения, а также риску смертности. Наследственные синдромы теломер (4, 5) являются высокоинформативными для выяснения ролей и взаимодействий дефектов механизма поддержания длины теломер в старении представителей обычных популяций человека и возрастных болезнях. Ухудшение работы поддерживающих длину теломер механизмов оказывает на клетки патофизиологическое влияние, эффекты которого могут предшествовать или взаимодействовать с определенными клеточными проявлениями старения (6). Так как последствия нарушенного поддержания длины теломер у человека проявляются специфично для различных типов клеток и тканей, они, соответственно, различаются при разных возрастных болезнях. В особенности в случае рака генетические детерминанты более длинных теломер повышают риски специфично для разных типов опухолей. Недавние достижения в понимании взаимосвязей между смертностью/возрастными болезнями и поддержанием длины теломер, в основе которых лежит генетические и негенетические факторы, выделяют важность ролей поддержания длины теломер в развитии болезней старения и их особенностях у человека. Биология теломер активно анализируется на модельных системах (1–4, 7–11).
В данной статье основное внимание уделяется человеческим генетическим и клиническим данным, имеющим отношение к вопросу о том, является ли малая длина теломер у человека побочным фактором или причиной развития болезней и синдромов старения. Согласно наиболее убедительному из современных представлений, укорочение теломер может как способствовать, так и быть результатом этиологии и прогрессии заболевания, а также в некоторых ситуациях может запускать порочный круг, взаимодействующий с другими лежащими в основе заболеваний процессами.
Утрата и восстановление теломер
Многие взрослые человеческие клетки, такие как фибробласты, имеют низкие или нерегистрируемые уровни теломеразы. В тканевой культуре такие клетки претерпевают прогрессивное укорочение теломер. Когда теломеры достигают критично короткой длины или подвергаются значительным повреждениям лишенные защиты теломеры запускают непрерывную передачу сигналов о повреждении ДНК. Это приводит к нарушению транскрипционных профилей и клетки вступают в фазу физиологического старения. В зависимости от типа клетки, характеристики фазы физиологического старения имеют различные последствия (рисунок 2) (11).
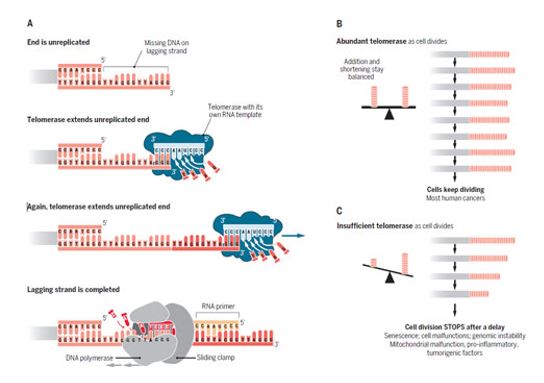
Рисунок 2. Для долгосрочного поддержания длины теломерной ДНК необходима теломераза. (А) Репликация теломерной ДНК. После удлинения одной цепочки ДНК с помощью обратнотранскриптазного механизма синтеза теломерной ДНК, осуществляемого теломеразой, следует удлинение второй цепочки теломерной ДНК, осуществляемое
У человека и модельных организмов (в том числе млекопитающих) теломерная ДНК часто особенно восприимчива к повреждениям и возникновению аномалий, появляющихся по всему геному.
Это может показаться парадоксальным, что теломеры, являющиеся частью генома, назначением которой является его защита, настолько подвержены повреждениям. Однако теломеры могут быть своего рода «службой экстренного реагирования» на угрозы стабильности генома и проблемы с поддержанием целостности ДНК, а теломерная ДНК выступает в роли «тревожного звонка», изменяющего реакции клетки до того, как произошло повреждение последовательности, кодирующей генетическую информацию.
В целом, несмотря на то, что клетках большинства человеческих тканей теломеры укорачиваются на протяжении всей жизни, идея о непрерывно тикающих митотических часах тоже чрезмерно упрощена. Помимо прочих факторов она сильно усложняется варьирующими уровнями теломеразной активности и, соответственно, разными возможностями увеличения длины теломер в стволовых клетках. Они способны непрерывно обновлять клетки соматических тканей. Например, высокие уровни теломеразы характерны для гемопоэтических стволовых клеток и стволовых клеток кишечных ворсинок, их промежуточных делящихся
Регулирование теломер чрезвычайно интерактивно
Человеческая теломерная ДНК формирует каркас для иерархии белков, варьирующих от нуклеосомных гистонов до шелтериновых компонентов и ассоциированных с определенными условиями факторов восстановления повреждений ДНК (рисунок 1) (1). Увеличение длины теломер и регуляция этого процесса являются частью обширных сетей клеточных взаимодействий. Они включают плотное регулирование экспрессии и активности теломеразы, а также комплекса защищающих теломеры белков, известных как шелтерины. Помимо защиты теломер от разрушительного действия процессов реагирования на повреждения ДНК (1), шелтериновым компонентам принадлежит двоякая роль: они как привлекают теломеразу к теломерам, так и предотвращают ее воздействие на них чрезвычайно отрегулированной манере. Другие факторы, в том числе белки восстанавливающие повреждения ДНК, также наносят теломерам кратковременные визиты, часть которых осуществляется посредством специфичных к шелтериновым компонентам взаимодействий. Помимо поддержания длины теломер шелтериновые компоненты выполняют и другие функции (врезка 1). Таким образом им принадлежат тонко сбалансированные роли и гораздо более важен динамично регулируемый баланс шелтериновых компонентов, чем их большое количество.
Старение человека по сравнению с модельными организмами
Помимо очевидных проявлений старения у человека, таких как седые волосы, морщинистая покрытая пигментными пятнами кожа, атрофия мышечной ткани и изменения характера жировых отложений, на протяжении последних десятилетий жизни человека значительно увеличивает восприимчивость к различным заболеваниям. К таким возрастным состояниям относятся угасание иммунной функции,
Продолжительность жизни эукариот варьирует в пределах более чем пяти порядков. Перенос результатов, полученных при работе с лабораторными модельными системами, может быть весьма проблематичным из продолжительного многодекадного временного интервала человеческого старения, а также различий, таких как масса тела и других эволюционных отличий. Теломеры укорачиваются на протяжении всей продолжительность жизни человека, в том числе в течение периода старения. В противоположность этому, при нормальном старении у большинства используемых для лабораторного изучения старения животных моделей со значительно более короткой продолжительностью жизни практически не происходит укорочения теломер. Так (за исключение случаев когда, возможность поддержания длины теломер устранена генетическими методами) лабораторные мыши и крысы обычно умирают в преклонном возрасте, имея интактные относительно длинные теломеры (13), также как и другие широко изучаемые короткоживущие модели (не относящиеся к млекопитающим), такие как круглые черви Caenorhabditis elegans (9), полосатый данио (zebrafish) (14) и африканский нотобранх Фунцера (African turquoise killifish) (8). В процессе эволюции опосредуемая теломеразой система поддержания длины теломер была полностью утрачена плодовыми
Врезка 1. Обнаруженные недавно функции шелтериновых компонентов, не касающиеся теломер
Различные шелтериновые компоненты обнаруживаются как по всему объему ядра, так и на теломерах. Некоторые из них являются факторами транскрипции и оказывают свое действие по всему геному. Многочисленные шелтериновые компоненты, начиная от почкующихся дрожжей и заканчивая млекопитающими (и, предположительно, человеком), демонстрируют в высшей степени регулируемое связывание с огромным количеством нетеломерных регионов генома, где они регулируют транскрипцию ряда систем генов, специфичных для типа и стадии развития клетки, например, сохранившийся в ходе эволюции шелтерин RAP1 имеет транскрипционные роли. Генетические манипуляции над RAP1 у мышей вызывают развитие специфичного для пола ожирения даже в отсутствие повреждений теломер. Таким образом, другие системы генов, помимо регулирующих реакции на повреждение ДНК и стресс, регулируются шелтериновыми компонентами, выступающими в роли факторов транскрипции, и могут подвергаться влиянию изменений соотношения их содержания на теломерах и в других регионах генома (61).
Сами шелтерины, в свою очередь, управляются механизмами регуляции экспрессии и модификации генов, специфичными для типа и стадии развития клетки. Например, хорошо изученный способствующий долголетию фактор транскрипции FOXO3 повышает экспрессию важного защищающего теломеры белка POT1a в нервных стволовых клетках мышей. Такая регуляция может быть особенно важна для этих недавно выявленных ролей шелтериновых компонентов, не имеющих отношения к теломерам. Таким образом, влияние теломерных комплексов распространяется от транскрипционных реакций, вызываемых сигналами о повреждении ДНК при нарушении концевых участком теломер, до многофункциональных шелтериновых компонентов, выступающих в роли факторов транскрипции, необходимых для репрограммирования систем, в состав которых входят гены метаболизма (61).
Эти многочисленные уровни открывают сложнейшие двусторонние диалоги между целостностью теломер и клеточными и организменными функциями. Судя по всему, их взаимодействие в многочисленных тканях и на различных этапах жизни человека носит исключительно сложный характер.
Генетика: нарушение теломер может приводить к болезням
То, что неадекватное поддержание длины теломер может вызывать у лабораторных модельных организмов развитие некоторых фенотипов старения эукариот, продемонстрировано только путем экспериментального удаления генов, обеспечивающих поддержание длины теломер (4, 8, 9, 13, 16, 17). Например, у мышей полностью нулевой генотип компонента теломеразы или защитного белка теломер после достаточного для утраты защиты укорочения теломер приводит к появлению характерных для ускоренного старения фенотипов (4, 13). Сложность заключается в выяснении степени, до которой недостаточность поддержания длины теломер у человека приводит к запуску этих же патологических механизмов в процессе нормального старения.
Моногенетические наследственные нарушения поддержания длины теломер на простейшем концептуальном уровне четко демонстрируют, что лишенным защиты теломерам могут принадлежать причинные роли в старении и развитии возрастных болезней у человека. Такие нарушения вызываются менделевскими мутациями, нарушающими функционирование теломер. Они обычно (но не всегда) манифестируют in vivo (в живом организме) как очень короткие теломеры. Это является результатом чрезмерного укорочения теломер с последующей утратой их концевых участков и защиты. Как описано выше, запускаемые поврежденными теломерами сигналы вызывают появление фенотипов, ведущих к развитию заболеваний.
Такие инактивирующие мутации отдельных генов на сегодняшний день известны для 11 генов человека. Каждый мутировавший ген выполняет известную, молекулярно описанную, непосредственную функцию в поддержании длины теломер: кодирует один их компонентов теломеразы (TERC, TERT, DKC1, NOP10, NHP2 или WRAP53) или связывающий теломеры белок, обнаруживаемый на теломерах in vivo и выполняющий экспериментально доказанную функцию защиты теломер (TINF2, RTEL1, POT1, CTC1 или TPP1) (4, 5).
Эти наследственные нарушения представляют практически бесконечное количество ткане- и органоспецифичных патологий. Их все объединяет один общий причинный молекулярный механизм: реакция на незащищенные (обычно укороченные до критичной длины) теломеры. Они объединяют множество фенотипов экспериментальных мышиных моделей, не имеющих генов, необходимых для поддержания длины теломер (18–20). Эта коллекция клинически разнородных патологий, вызванных моногенетическими мутациями, известна как «наследственные теломерные синдромы». Эти заболевания часто имеют аутосомно доминантный характер. У человека функциональная гаплоидентичность часто обуславливает патологию и дозу гена (количество аллелей локуса в генотипе) и, соответственно, уровень соответствующего продукта гена, входящего в механизм поддержания длины теломер, важен для защиты от широкого спектра заболеваний и синдромов, поражающих весь организм.
У пациентов наблюдаются клиническая вариабельность, неполная пенетрантность и вариабельная экспрессивность мутации (5). Несмотря на это, все более очевидно, что наследуемые теломерные синдромы имеют характерные закономерности. Они могут включать один или несколько из следующих параметров: утрата иммунной функции
Мышиные модели недостаточности теломеразы обычно требуют нескольких поколений для того, чтобы их прогрессивно укорачивающиеся теломеры достигли точки манифестации фенотипов старения (4, 8, 9, 13, 16). Аналогичным образом отличительным признаком человеческих теломерных синдромов является генетическая антиципация, при которой последующие поколения носителей мутации в семейной генеалогии последовательно демонстрируют более раннее проявление заболевания, при этом типы заболеваний также характерным образом отличаются для последующих поколений. В особенности для поздних поколей характерна ранняя смерть. В совокупности с мышиными моделями эти наблюдения легли в основу революционных взгляда на вопрос: доказательство того, что сами по себе короткие теломеры являются основной механистической причиной развития фенотипов заболевания. Удивительным образом заболевание манифестирует даже у не являющихся носителями мутации потомков, если они наследуют короткие теломеры от пораженного заболеванием родителя, являющегося носителем мутации (4, 5, 21).
Теломеры также оказывают
Врезка 2. Совокупные/синергичные эффекты укорочения теломер и генов специфичных заболеваний
В мышиных моделях с двойными Wrn−/− Terc−/− (62) или тройными Wrn−/− BlmM3/M3 Terc−/− мутациями (23) фенотипы делеции теломеразы проявляются у более ранних поколений после делеции TERC, по сравнению с шестым поколением для модели на основе животных дикого типа с мутацией Terc−/− (23, 62).
Дополнительные фенотипы, специфичные для человеческих синдромов Вернера и Блума, также наблюдаются и имеют более тяжелый характер, чем характерно для мышей линий Wrn−/− или Wrn−/− BlmM3/M3 с TERC дикого типа. Недавнее исследование in vitro («в пробирке») показало, что добавление теломеразы защищает человеческие стволовые клетки с синдромом Вернера от преждевременного старения (24), что подкрепляет более раннее наблюдение, согласно которому нарушение функций теломер способствует формированию фенотипа старения при синдроме Вернера (63).
У предрасположенных к сахарному диабету мышей линии Akita, имеющих мутацию Ins2C96Y/WT в гене инсулина, неправильное сворачивание молекулы инсулина вызывает стресс эндоплазматического ретикулума и приводит к апоптозу
бета-клеток поджелудочной железы (25). Деления TERC у мышей линии Akita ведет к усилению апоптозабета-клеток более выраженной утрате толерантности к глюкозе по сравнению с наличием только мутации Ins2C96Y/WT. Это свидетельствует о совокупном влиянии нарушения функций теломер и стресса эндоплазматического ретикулума.Мышечная дистрофия Дюшенна является приводящим к атрофии мышечной ткани заболеванием, вызываемым мутацией гена дистрофина, приводящей к дегенерации мышечной ткани и преждевременной смерти. Мышиная модель с мутацией, аналогичной человеческой, демонстрирует неожиданно слабовыраженные фенотипы. Однако у мышей с двойной мутацией, для которых характерно отсутствие дистрофина и короткие теломеры (mdx/mTRKO), развиваются фенотипы, аналогичные человеческим, в том числе тяжелые функциональные нарушения работы сердца, такие как дилатация желудочка, нарушения сократимости и проводимости, а также ускоренная смертность (64). Эти нарушения сердечной деятельности сопровождаются эрозией теломер. Длина теломер кардиомиоцитов у второго поколения (G2) мышей линии mdx/mTRKO меньше длины теломер мышей второго поколения мышей с нокаутированным mTR, что свидетельствует об ускорении процесса укорочения теломер в присутствии мутации гена дистрофина.
В данном случае синергичный эффект также наблюдается как для теломер, так и для мышечной дистрофии Дюшенна.
Эти данные расширяют спектр возможного влияния укорочения теломер на старение и заболевания и подчеркивает то, что часто «действие проявляется во взаимодействии».
Фактор, потенцирующий заболевания и прогнозирующий смертность
Несмотря на то, что наследственные теломерные синдромы часто манифестируют раньше и более тяжелых формах, их фенотипы соответствуют фенотипам заболеваний, частота развития которых в общей популяции человека резко возрастает с возрастом. Многие из патологий, ассоциированных с нормальным старением человека, в том числе угасание иммунной функции (26), рак (27, 28), сахарный диабет (29) и заболевания
Среднее значение длины теломер лейкоцитов или мононуклеарных клеток периферической крови является отражением системного влияния поддержания длины теломер в других тканях и, что очень важно, доли вступивших в фазу физиологического старения циркулирующих в кровотоке клеток иммунной системы. Существует множество задокументированных доказательств роли воспаления в патологической прогрессии таких заболеваний, как болезни
Большинство человеческих болезней старения подвергается влиянию сложных генетических и негенетических факторов. Примеры мышиных моделей (врезка 2) демонстрируют, что приписываемая заболеванию причинная обусловленность может быть неполной без принятия во внимание малой длины теломер. Объективные полногеномные исследования ассоциаций в локусах, влияющих на длину теломер лейкоцитов человека неизменно указывали на локусы, содержащие известные гены, кодирующие белки, защищающие теломеразу или теломеры. Менделевская рандомизация выявила причинную взаимосвязь между малой длиной теломер лейкоцитов и повышенным риском развития ишемической болезни сердца при проведении одного из таких крупных европейских исследований (31). Это позволило определить сеть главных вариантов аллелей распространенных генов, вместе объясняющих примерно 1% общей вариабельности длины теломер лейкоцитов. Пять наиболее важных (TERC, TERT, NAF1, OBFC1 и RTEL1) из этих семи генов кодируют компоненты, оказывающие молекулярно описанное непосредственное действие на теломеры. Количество аллелей этих семи важных генов, ассоциированных с более короткими теломерами, обеспечивали дополнительное прогнозирование риска развития ишемической болезни сердца. Риск развития ишемической болезни сердца были примерно на 20% выше среднестатистического у индивидуумов с наиболее высоким показателем для аллелей шести из упомянутых семи наиболее важных генетических вариантов, ассоциированных с короткими теломерами (31). Также была продемонстрирована ассоциация между ишемической болезнью сердца и распространенными единичными нуклеотидными полиморфизмами («снипами») генов NAF1 и OBF1C (32, 33).
По мере старения человека происходит уменьшение средней длины теломер и рост смертности. Хронологический возраст отвечает менее чем на 10% вариабельности длины теломер человека. Недавние крупные когортные исследования прояснили, что, независимо от возраста и других известных ранее факторов риска смертности, степень укорочения теломр также является четким статистическим прогностическим фактором человеческой смертности от любых причин (36, 37). Однако поддержание длины теломер является в высшей степени интерактивным и, как описано ниже, измерение длины теломер демонстрирует значительно более высокую прогностическую мощность при использовании в комбинации с другими факторами.
Недавно проведенное в Калифорнии исследование с участием 100 000 взрослых людей, согласно результатом которого 75 лет является возрастом максимального риска смертности, показало, что у людей старше 75 лет тенденция к укорочению теломер с возрастом разворачивается в обратную сторону (38). Эта
В большинстве случаев смертность в крупных когортах, использованных до настоящего времени для изучения длины теломер лейкоцитов, была результатом распространенных болезней старения, преимущественно
Возможность использования короткой длины теломер как прогностического фактора для развития болезней в будущем согласуется с предположением о причинной роли укорочения теломер, но не является его доказательством. Доказательство частичного вклада малой длины теломер было получено в описанных выше полногеномных исследованиях ассоциаций, посвященных изучению ишемической болезни сердца. С биологической точки зрения это согласуется с фенотипами болезни и смертности, характерными для пациентов с наследственными теломерными синдромами и мышей, не имеющих гена, необходимого для поддержания длины теломер.
Разные виды рака: теломераза балансирует на острие
В опухолевых клетках, часто ведущих себя как бессмертные, поддержание длины теломер обеспечивается различными способами. В 80–90% полностью злокачественных опухолей человека активность теломеразы раковых клеток повышена по сравнению с нормальными клетками соответствующих тканей. Однако в основе развития более 200 типов рака человека лежит широкий спектр этиологий рака и последовательностей событий. Как и следует ожидать с учетом этого разнообразия, разные типы рака отличаются тем, как повышенная способность к поддержанию длины теломер или ее компрометация стимулирует сложные процессы развития и прогрессии рака. С одной стороны, наследственные теломерные синдромы демонстрируют, что в масштабах организма неадекватная активность теломеразы приводит к повышению частоты развития опухолей системы крови, плоских клеток кожи и
Негенетические факторы: внешние стрессоры и образ жизни
Коэффициент наследуемости для длины теломер человека варьирует в диапазоне 30–80%. Очень короткие теломеры наследуются в семьях с мутациями, вызывающими наследственный теломерный синдром (21). Также было высказано предположение, что в общей популяции, помимо обычных наследуемых генетических факторов, непосредственное наследование теломер через родительские гаметы может отвечать за значительную часть оценочной наследуемости длины теломер (44).
Однако наследуемость длины теломер снижается с возрастом (45), тогда как были продемонстрированы выраженные негенетические влияния, в особенности воздействующие на всех факторы окружающей среды (46). Эпигенетические влияния на длину теломер могут в поиске связующих звеньев для большого количества рассматриваемых в настоящее время негенетических факторов внешней среды и других факторов, оказывающих влияние на поддержание длины теломер. При изолированном рассмотрении только увеличивающих длину теломер аллелей трех генов, обеспечивающих поддержание длины теломер, оказалось, что все они повышали риск смертности от всех типов рака (36). Однако выявлении более длинных теломер было ассоциировано со сниженной смертностью от всех типов рака. Следовательно влияние генетических и негенетических факторов, обеспечивающих бОльшую длину теломер, различается для разных типов рака (рисунок 3). Поэтому негенетические, в том числе эпигенетические, влияния на поддержание длины теломер представляют большой интерес с точки зрения их роли в этиологии заболеваний.
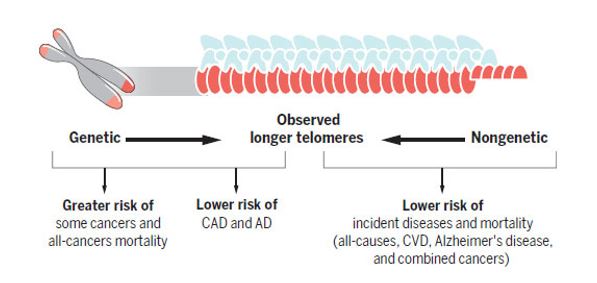
Рисунок 3. Различные влияния на поддержание длины теломер имеют болезнеспецифичные последствия. Наблюдаемые изменения длины теломер, происходящие под действием комбинаций генетических и негенетических влияний. Более длинные теломеры ассоциированы с более низкими общими рисками смертности и заболеваемости. Варианты специфичных генов, способствующие поддержанию большей длины теломер, снижают риски развития ишемической болезни сердца и болезни Альцгеймера, одновременно повышая риск развития определенных типов рака и общей смертности от рака. Негенетические влияния, обеспечивающие более эффективное поддержание длины теломе, имеют защитный характер.
У человека ассоциации между стрессом и длиной теломер можно увидеть на ранних этапах жизни. Длина теломер новорожденных была меньше пропорционально уровням стрессов, перенесенных матерями во врем беременности (47). Более высоки степени подверженности насилию или пренебрежению в детстве, а также различным категориям негативного детского опыта были ассоциированы со значительно более короткими теломерами при измерении как у детей, так и ретроспективно — у взрослых (48). «Дозозависимые» эффекты воздействия стрессоров на длину теломер также наблюдались у взрослых. В качестве примеров можно привести продолжительность периодов подверженности домашнему насилию, нелеченой депрессии или ухода за членом семьи, страдающим от хронического заболевания (49). Экспериментальные исследования, в рамках которых молодых птиц подвергали стрессовым воздействиям, продемонстрировали, что хронический психологический стресс является причинным фактором укорочения теломер (50).
Механизмы взаимосвязи между стрессом и длиной теломер в настоящее время начинают изучаться и, скорее всего, являются многочисленными. Передача материнского стресса потомкам может быть опосредована прямыми эффектами, такими как меньшая длина теломер родительских гамет, эпигенетика, или непрямыми эффектами, например, изменением внутриматочного окружения
Подобные исследования предоставляют наводящие на размышления свидетельства в пользу того, что хронический психологический стресс может быть одним из причинных факторов укорочения теломер у человека. Взрослые люди с тяжелыми депрессивными заболеваниями демонстрируют тенденцию к укорочению теломер (54), в особенности при большей тяжести и продолжительности депрессии (55). Таким образом малая длина теломер с большой вероятностью является результатом развития заболевания, хотя их укорочение может и предшествовать манифестации депрессии у детей, входящих в группу высокого риска (52).
Другие факторы, считающиеся ассоциированными с длиной теломер, варьируют от социальных и экологических факторов до факторов образа жизни, таких как курение и физические нагрузки (49, 56). Несмотря на то, что длина теломер или ассоциированных с теломерами аллель сами по себе оказывают небольшое влияние на заболевание, это влияние может быть приумножено, например, депрессией (57) или курением (58, 59). Таким образом, интерактивное влияние негенетических и генетических детерминант теломер является потенциально мощным и сравнительно плохо изученным.
Важные для клиники результаты и выводы
Единый механизм — утрата защитной функции теломер — вызывает развитие наследственных теломерных синдромов, приводящих к патологиям, схожим с возрастными болезнями. Их системная (поражающая весь организм) природа четко указывает на то, что при часто сопутствующих друг другу возрастных болезнях (таких как сахарный диабет и
Рисунок 4. Взаимосвязь между укорочением теломер и возрастными болезнями человека. Укорочение теломер изображено как основополагающий, повсеместный и интерактивный фактор, вовлеченный в этиологию старения и развития возрастных болезней. Так как он подвержен влиянию как негенетических так и генетических воздействий, поддержание длины теломер является поддающимся влиянию интерактивным индикаторов общего состояния здоровья.
За исключением редких теломерных синдромов, измерение длины теломер предоставляет только статистическую оценку вероятности и само по себе не является специфичным диагностическим параметром для индивидуума. Так как длина теломер подвержена влиянию такого множества негенетических факторов, результаты измерения уровня поддержания длины теломер могут быть использованы в качестве промежуточных показателей для оценки «экспозома», то есть совокупности всех воздействий (от англ. «exposures»), способствующих развитию болезни. Изучение взаимодействий между независимыми и перекрывающимися механизмами, влияющими на длину теломер человека, находится на ранних стадиях. В новой эре точных медицинских исследований важной задачей будет выяснение того, позволит ли комбинирование других прогностических факторов — геномных ассоциаций, клинических и поведенческих данных, а также данных о заболеваниях — с длиной теломер повысить точность прогнозирования прогресса и исходов заболеваний. Очень наглядный пример синергизма был получен при проведении исследования с участием когорты пациентов с раком мочевого пузыря. Наименьшее среднее значение длины теломер лейкоцитов, измеренное на момент постановки диагноза рака мочевого пузыря, само по себе не было статистически значимо ассоциировано с будущей смертностью. Однако при комбинировании с поставленным диагнозом депрессии медиана времени выживания уменьшилась с 200 месяцев (для всех других групп) до 31 месяца при проведении многовариантного анализа с поправками на демографические и клинические переменные (57).
Поддержание длины теломер у человека представляет собой неожиданно тонкий баланс между снижением риска развития множества возрастных болезней и одновременным повышением вероятности возникновения определенных менее распространенных, но часто летальных типов рака. Поэтому не имеющие клинического одобрения лекарственные средства, предназначенные для повышения активности теломеразы, вполне способны повысить долгосрочный риск развития рака. Биологию теломер лучше всего рассматривать в следующем контекст: она подает надежду как мощный интерактивный фактор, который может быть полезен в точной медицине для клинического мониторинга состояния здоровья и оценки развития заболевания. Выяснение того, каким образом генетические и негенетические детерминанты механизма поддержания длины теломер могут взаимодействовать (между собой и с другими причинами развития болезней) для того, чтобы стать ограничивающими для рисков развития заболеваний, требует проведения дальнейших исследований. Ранние эмпирические данные, полученные в исследованиях с участием людей, указывают на то, что здоровый образ жизни может сглаживать влияние стресса или депрессии на длину теломер (56), а также на то, что поведенческие вмешательства могут повышать эффективность поддержания длины теломер в определенных условиях (60). Продолжающиеся механистические исследования улучшат понимание вопроса пластичности механизма поддержания длины теломер и позволят установить, когда и каким образом вмешательства могут эффективно оказывать влияние на развитие заболеваний и состояние здоровья.
Использованные источники:
1. 1. A. Sfeir, T. de Lange, Science 336, 593–597 (2012).2.
3. Z. Xie et al., Cell 160, 928–939 (2015).
4. M. Armanios,
5. G. Glousker, F. Touzot, P. Revy, Y. Tzfati,
6. E. Sahin et al., Nature 470, 359–365 (2011).
7.
8. I. Harel et al., Cell 160, 1013–1026 (2015).
9. B. Meier et al., PLOS Genet. 2, e18 (2006).
10. W. Palm, T. de Lange, Annu. Rev. Genet. 42, 301–334 (2008).
11. Simm, J. Campisi, Exp. Gerontol. 59, 1–2 (2014).
12. S. Petersen, G. Saretzki, T. von Zglinicki, Exp. Cell Res. 239, 152–160 (1998).
13.
14. M. Anchelin et al., Dis. Model. Mech. 6, 1101–1112 (2013).
15.
16. P. Missios et al., Nat. Commun. 5, 4924 (2014).
17.
18. M. Armanios, J. Clin. Invest. 123, 996–1002 (2013).
19. T. Vulliamy et al., Nature 413, 432–435 (2001).
20.
21.
22.
23. X. Du et al., Mol. Cell. Biol. 24, 8437–8446 (2004).
24.
25. N. Guo et al., PLOS ONE 6, e17858 (2011).
26. S. Cohen et al., JAMA 309, 699–705 (2013).
27.
28. H. Ma et al., PLOS ONE 6, e20466 (2011).
29. J. Zhao, K. Miao, H. Wang, H. Ding,
30.
31.
32. H. Ding et al., Clin. Interv. Aging 9, 857–861 (2014).
33.
34. Y. Zhan et al., JAMA Neurol. 72, 1202–1203 (2015).
35.
36. L. Rode,
37.
38. K. Lapham et al., Genetics 200, 1061–1072 (2015).
39.
40. P. Willeit, J. Willeit, A.
41.
42. S. Horn et al., Science 339, 959–961 (2013).
43. K. Nexerova,
44. T. De Meyer et al., Eur. J. Hum. Genet. 22, 10–11 (2014).
45.
46.
47. S. Entringer et al., Proc. Natl. Acad. Sci. U.S.A. 108,
48.
49. J. Lin, E. Epel, E. Blackburn, Mutat. Res. 730, 85–89 (2012).
50.
51.
52. H. Gotlib et al., Mol. Psychiatry 20, 615–620 (2015).
53. J. Choi,
54.
55. D. Lindqvist et al., Neurosci. Biobehav. Rev. 55, 333–364 (2015).
56. E. Puterman, J. Lin, J. Krauss,
57. Lin et al., Cancer Epidemiol. Biomarkers Prev. 24, 336–343 (2015).
58. Gu et al., Cancer Prev. Res. (Phila.) 4, 514–521 (2011).
59. J. Raschenberger et al., Sci Rep 5, 11887 (2015).
60.
61. J. Ye,
62. S. Chang et al., Nat. Genet. 36, 877–882 (2004).
63. L. Crabbe, A. Jauch,
64. F. Mourkioti et al., Nat. Cell Biol. 15, 895–904 (2013).
ОБЗОР
Elizabeth H. Blackburn, Elissa S. Epel, Jue Lin



